Enzyme
Evolution:
Recent scientific studies provide strong evidence
that nucleic acid polymerases share a common ancestor. Structural analyses support
the notion that the similarities between polymerases go beyond the arbitrary assignment
into classes on the basis of the use of DNA or RNA templates and deoxyribonucleoside or
ribonucleoside triphosphates (Figure 18).
In fact, it has been postulated that most of the nucleic acid polymerizes belong to a
polymerase superfamily containing closely related active sites that are similarly
positioned within the polymerase active cleft (Figure 19).
Thus, it appears that there is a genetic polymerase module that provide
the active site the architecture to carry out the phosphoryl transfer reaction (Joyce, 1997). Only subtle modifications to this
module achieve the substrate specificity that is unique for each polymerase class. We see over and over in the course of evolution the tendency of
nature to copy and paste particular structural motifs.
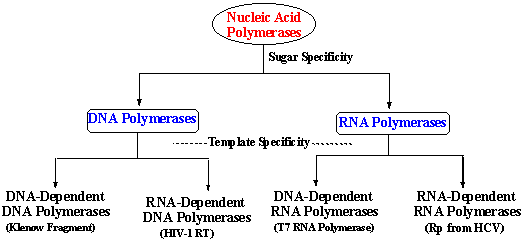
Figure 18. Schematic representation
illustrating the division into classes of nucleic acid polymerases in terms of sugar and
template specificities.
As Joyce
C. (1997) has stated "nucleic acid polymerases are suffering from an
identify crisis." Recent published papers have brought us, a
DNA polymerase as a reverse trancriptase (Ricchetti
et. al., 1993), the T7 RNA polymerase as a DNA polymerase (Sousa el. al., 1995), and a DNA polymerase as an
RNA polymerase (Gao et. al., 1997). This
switch in specificity was done by single amino acid substitution. As illustrated in Figure 19, the polymerase domains of the Klenow
fragment, the HIV-1 RT, and that for T7 RNA polymerase are structurally similar. The
yellow spheres shown in the palm domain represent the catalytic aspartate residues. The relative positions of the residues involved in sugar
discrimination are indicated in each polymerase structure.
Figure 19. Illustration of the
polymerase domains of Klenow fragment, HIV-1 RT, and T7 RNA polymerase (Joyce, 1997).
DNA polymerases discriminates
against ribonucleotide through a "steric gate" mechanism. For example, the
Klenow fragment and the HIV-RT polymerase discriminate against ribose sugar because bulky
amino acid residues (Phe762 and Tyr155, respectively) make steric interaction with the
2'-OH group of the incoming nucleotide (see Figure 19). The Moloney murine leukemia
virus (MoMLV) polymerase contains Phe155, which is homolog to Tyr155 in HIV-RT. Figure 20 shows how Phe155 in
MoMLV clashes with the 2'-OH group of the incoming ribonucleotide (Gao et. al., 1997). Thus, mutation of this
bulky residue to alanine, as it was found, will switch the sugar specificity of the
polymerase.
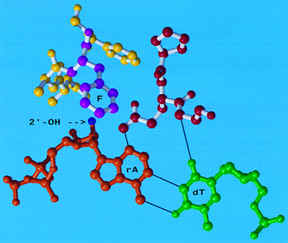
Figure 20. In MoMLV RT, Phe155 acts
as a "steric gate" to prevent incorporation of r-NTPs (Gao et. al., 1997).
On the other hand, T7 RNA polymerase uses a totally
different mechanism to discriminate against deoxyribose nucleotides. Studies have
shown that, in T7 RNA polymerase, Tyr639 makes hydrogen-bonding interaction with the
2'-OH group of the incoming ribonucleotide. Mutation in this position from Tyr to
Phe renders the polymerase specific for deoxynucleotides. (Sousa et. al., 1995).
Bacteriophage T7 RNA polymerase
shares some amino acid sequence similarities with other RNA polymerases. Table 2
illustrates the result obtained when the FASTA program was used
for sequence alignment. As it is shown, only bacteriophage T3 RNA polymerase has the
highest amino acid identity (82 %) to T7 RNA polymerase, while the other RNA polymerases
share only about a 29 % amino acid identity.
Table 2. Amino acid identity
between T7 RNA polymerases and other RNA polymerases.
B 10 20 30 40 50 60
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 1 GKPLG-PSGLKWLKIYGANLF--GLDKKSFDERIA--WVEENLDNIIDSANNPLTGE-WW 54
1QLN_A 449 GKPIG-KEGYYWLKIHGANCA--GVDKVPFPERIK--FIEENHENIMACAKSPLENT-WW 502
gi 133451 440 GRPVNgVEALKWFCINGANLW--GWDKKTFDVRVSnvLDEEFQDMCRDIAADPLTFT-QW 496
gi 3914832 584 GKPLG-KSGLRWLKIHIANLYagGVDKLAYEDRIA--FTESHLEDIFDSSDRPLEGKrWW 640
gi 3914826 601 GRPLG-KSGLHWLKIHLANLYagGVEKLSHDARLA--FVENHLDDIMDSAENPIHGKrWW 657
gi 3914825 696 GKPLG-PKGLNWLKVHLANLF--GISKKDFATRQA--FVDDNMQEVFDSADRPLDGNkWW 750
gi 133457 854 GKKLG-PSGLKWLKIHLSNLF--GFDKLPLKDRVA--FTESHLQDIKDSAEKSLTGDrWW 908
gi 730615 809 GKPLG-ESGLRWLKVHLANVY--GFDKASLQERQD--FADENIENIRDSVNNPLNGNqWW 863
gi 3914823 830 GRPLG-PHGLDWLKIHLVNLTg-LKKREPLRKRLA--FAEEVMDDILDSADQPLTGRkWW 885
|
70 80 90 100 110 120
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 55 DKADNPFQFLAFCFELAKYLDNPd---pESFISHLPIHQDASCNGLQHYAALLRDSVLAK 111
1QLN_A 503 AEQDSPFCFLAFCFEYAGVQHHG-----LSYNCSLPLAFDGSCSGIQHFSAMLRDEVGGR 557
gi 133451 497 AKADAPYEFLAWCFEYAQYLDLVdegraDEFRTHLPVHQDGSCSGIQHYSAMLRDEVGAK 556
gi 3914832 641 LNAEDPFQCLAACINLSEALRSPf---pEAAISHIPIHQDGSCNGLQHYAALGRDKLGAD 697
gi 3914826 658 LKAEDPFQCLAACVILTQALKSPs---pYSVISHLPIHQDGSCNGLQHYAALGRDSFEAA 714
gi 3914825 751 SKADDPFQALAACFEIAEAVRSGd---hESYISHIPIQQDGTCNGLQHYAALGGDIEGAK 807
gi 133457 909 TTADKPWQALATCFELNEVMKMDn---pEEFISHQPVHQDGTCNGLQHYAALGGDVEGAT 965
gi 730615 864 LQAEDPWQCLATCFELAAALELEd---pTKYVSHLPIHQDGTCNGLQHYAALGGDTWGAQ 920
gi 3914823 886 MGAEEPWQTLACCMEVANAVRASd---pAAYVSHLPVHQDGSCNGLQHYAALGRDSVGAA 942
|
130 140 150 160 170 180
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 112 AVNLLPStDSPRDFYSFVAPCVIKFLKADAEKK------------T-------------- 145
1QLN_A 558 AVNLLPS-ETVQDIYGIVAKKVNEILQADAINGtdnevvtvtdenTgeise--------- 607
gi 133451 557 AVNLKPS-DAPQDIYGAVAQVVIKKNALYMDADdattftsgsvtlSg------------- 602
gi 3914832 698 AVNLVTG-EKPADVYTEIAARVLKIMQQDAEEdp----------eT-------------- 732
gi 3914826 715 AVNLVAG-EKPADVYSEISRRVHEIMKKDSSKdp----------eS-------------- 749
gi 3914825 808 QVNLWPS-DHPSDVYEAVAEIVRGFLKKDAEAG--------------------------- 839
gi 133457 966 QVNLVPS-DKPQDVYAHVARLVQKRLEIAAEKG--------------------------- 997
gi 730615 921 QVNLVPG-DRPADVYSAVAKLVIKGIEDDLAK---------------------------- 951
gi 3914823 943 SVNLEPS-DVPQDVYSGVAAQVEVFRRQDAQR---------------------------- 973
|
190 200 210 220 230 240
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 146 -------KDIESAARLLDIGITRKVVKKTVMTIPYGVTYVGLRKQIEEKLEEKSDDEDEK 198
1QLN_A 608 ---kvklGTKALAGQWLAYGVTRSVTKRSVMTLAYGSKEFGFRQQVLEDTIQPAIDSGKG 664
gi 133451 603 ------tELRAMASAWDSIGITRSLTKKPVMTLPYGSTRLTCRESVIDYIVDLEEKEAQK 656
gi 3914832 733 -------FPNATYAKLMLDQVDRKLVKQTVMTSVYGVTYSGARDQIKKRLKERGTFEDDS 785
gi 3914826 750 -------NPTAALAKILITQVDRKLVKQTVMTSVYGVTYVGAREQIKRRLEEKGVITDER 802
gi 3914825 840 ----------DEMANFLKDKVTRSVVKPTVMTNVYGVTYVGARKQISEKLENIDGMEKLK 889
gi 133457 998 ----------DENAKILKDKITRKVVKQTVMTNVYGVTYVGATFQIAKQLSPIFDDRKES 1047
gi 730615 952 ---------DNEFAKAMHGKITRKVVKQTVMTNVYGVTYVGARKQVLKQIEAAYPNITAE 1002
gi 3914823 974 --------GMRVAQVLES-FITRKVVKQTVMTVVYGVTRYGGRLQIEKRLRELSDFPQEF 1024
|
250 260 270 280 290 300
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 199 LIF---VG---AATYLAKKVFKALREMFLLAKAIYNWLGACAKKLASLLTyyNSIVSIKL 252
1QLN_A 665 LMF---TQpnqAAGYMAKLIWESVSVTVVAAVEAMNWLKSAAKLLAAEVK--DKKTGEIL 719
gi 133451 657 AVA---EG----RTANKVHPFEDDRQDYLTPGAAYNYMTALIWPSISEVVkaPIVAMKMI 709
gi 3914832 786 LTF---H----ASCYAAKITLKALEEMFEAARAIKSWFGDCAKIIASENN---------- 828
gi 3914826 803 MLF---A----AACYSAKVTLAALGEIFEAARAIMSWLGDCAKIIASDNH---------- 845
gi 3914825 890 VAD---Y-----ANYLTKKVFEALRSLFTQAHEIQDWLSACCNLITHSLPadYIKEGIK- 940
gi 133457 1048 LDF---S------KYLTKHVFSAIRELFHSAHLIQDWLGESAKRISKSIRldVDEKSFKN 1098
gi 730615 1003 SGI---EAa-lLASYVTQHIFRAMSTMFKGAHDIQNWLGEIGGRVCRALTpeQLDEFERS 1058
gi 3914823 1025 VWE---A-----SHYLVRQVFKSLQEMFSGTRAIQHWLTESARLISHMGS---------- 1066
|
310 320 330 340 350 360
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 253 ----------------------------------KLDIPVIWVTPLGLPVVQPYRKSKKK 278
1QLN_A 720 ----------------------------------RKRCAVHWVTPDGFPVWQEYKKPIQT 745
gi 133451 710 rqla--------------------------rfaaKRNEGLMYTLPTGFILEQKIMATEML 743
gi 3914832 829 --------------------------------------AVCWTTPLGLPVVQPYRKPGRH 850
gi 3914826 846 --------------------------------------PVRWITPLGLPVVQPYCRSERH 867
gi 3914825 941 ----------------------------------DELTPVVWTTLLNLPIVQPYRNYKSR 966
gi 133457 1099 gn------------------------------kpDFMSSVIWTTPLGLPIVQPYREESKK 1128
gi 730615 1059 ersphgdgtasgenitlagnprkssahkndeilnNFQSTIIWTTPLRMPVVQPYRKHGTK 1118
gi 3914823 1067 --------------------------------------VVEWVTPLGVPVIQPYRLDSKV 1088
|
370 380 390 400 410 420
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 279 K-----TVPTNLQDTTLTLK-ETDKVDVR--KQKTAFMPNFIHSLDASHLILLADSCNKK 330
1QLN_A 746 RlnlmfLGQFRLQPTINTN--KDSEIDAH--KQESGIAPNFVHSQDGSHLRKTVVWAHEK 801
gi 133451 744 R-----VRTCLMGDIKMSLqvETDIVDEA--AMMGAAAPNFVHGHDASHLILTVCELVDK 796
gi 3914832 851 ------LVKTTLQVLTLSR--ETDKVMAR--RQMTAFAPNFIHSLDGSHMMMTAVACNRA 900
gi 3914826 868 ------LIRTSLQVLALQR--EGNTVDVR--KQRTAFPPNFVHSLDGTHMMMTAVACREA 917
gi 3914825 967 ------QIRTNLQTVFIEE--RDRTATVQphKQATAFPPNFIHSLDATHMFMTCLKCSEQ 1018
gi 133457 1129 ------QVETNLQTVFISDpfAVNPVNAR--RQKAGLPPNFIHSLDASHMLLSAAECGKQ 1180
gi 730615 1119 ------TVSTCMQDLVMTIpeRSDPVNRR--KQLQAFPPNFIHSLDASHMILSALHCDEL 1170
gi 3914823 1089 Kqi--gGGIQSITYTHNGD--ISRKPNTR--KQKNGFPPNFIHSLDSSHMMLTALHCYRK 1142
|
430 440 450 460 470 480
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 331 GG-L-NFASVHDCFGTHANDMDTLNKLLRETFIRLYSNDDYLLKLDDDFQNRIKNALDey 388
1QLN_A 802 YGiE-SFALIHDSFGTIPADAANLFKAVRETMVDTYESCDVLADFYDQFADQLHESQL-- 858
gi 133451 797 Gv-T-SIAVIHDSFGTHADNTLTLRVALKGQMVAMYIDGNALQKLLEEHEVRWMV----- 849
gi 3914832 901 G--L-SFAGVHDSFWTHACDVDVMNTILREKFVELYEK-PILENLLESFQKSFPD----- 951
gi 3914826 918 G--L-NFAGVHDSYWTHACDVDTMNRILREKFVELYNT-PILEDLLQSFQESYPN----- 968
gi 3914825 1019 N--I-NFAAVHDSYWTHACDVDQMNSLLREAFVLLHSN-NIMERLKQEFEERYKGFLVsk 1074
gi 133457 1181 G--L-DFASVHDSYWTHASDIDTMNVVLREQFIKLHEV-DLVLRLKEEFDQRYKNYVKig 1236
gi 730615 1171 G--L-TFAAVHDSFWTHASDIDSMNAVLRDAFIRIHSE-DVIGRLAAEFQARYKNSLYla 1226
gi 3914823 1143 G--L-TFVSVHDCYWTHAADVSVMNQVCREQFVRLHSE-PILQDLSRFLVKRFCSEPQki 1198
|
490 500 510 520 530 540
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 389 kenektde---------------------------------------------------- 396
1QLN_A ------------------------------------------------------------
gi 133451 ------------------------------------------------------------
gi 3914832 ------------------------------------------------------------
gi 3914826 ------------------------------------------------------------
gi 3914825 1075 kaikandedl-------------------------------------------------- 1084
gi 133457 1237 klkrstdlaqkiirirkdlsrklgrsttladeiyfekkrqelln---------------- 1280
gi 730615 1227 kietgtkvaqeiqrwrvrnklgprkelllekerqellrssnpedvergkkmispaslyel 1286
gi 3914823 1199 lea--------------------------------------------------------- 1201
|
550 560 570 580 590 600
....*....|....*....|....*....|....*....|....*....|....*....|
consensus ------------------------------------------------------------
1QLN_A ------------------------------------------------------------
gi 133451 ------------------------------------------------------------
gi 3914832 ------------------------------------------------------------
gi 3914826 ------------------------------------------------------------
gi 3914825 ------------------------------------------------------------
gi 133457 ------------------------------------------------------------
gi 730615 1287 yssaedltvpedlkevtignlagveetkvrrgremdeegevdgseeavehedgmhedeml 1346
gi 3914823 ------------------------------------------------------------
|
610 620 630 640 650 660
....*....|....*....|....*....|....*....|....*....|....*....|
consensus 397 ----------------------------------------eaitglsnydILKFPDLPKV 416
1QLN_A 859 ---------------------------------------------------DKMPALPAK 867
gi 133451 850 ---------------------------------------------------DTGIEVPEQ 858
gi 3914832 952 ---------------------------------------------------ISFPPLPER 960
gi 3914826 969 ---------------------------------------------------LVFPPVPKR 977
gi 3914825 1085 ----------------------------------------kakfgnksyipLEFPPLPAR 1104
gi 133457 1281 -----spliedrnvgekmvttvslfeditdldalelenggdensgmsvllpLRLPEIPPK 1335
gi 730615 1347 adeprdmdgnsgldelselrntnhfalsqkrakasiasggkqkhyldiwlpLVFPPIPEK 1406
gi 3914823 1202 -----------------------------------------------sqlkETLQAVPKP 1214
|
670
....*....|....*...
consensus 417 GDnTFDLKEILNSDYFFN 434
1QLN_A 868 GN--LNLRDILESDFAFA 883
gi 133451 859 GE--FDLNEIMDSEYVFA 874
gi 3914832 961 GD--FDLRKVLESTYFFN 976
gi 3914826 978 GD--FDLKEVLKSQYFFN 993
gi 3914825 1105 GA--LDLKKVLESKYFFS 1120
gi 133457 1336 GD--FDVTVLRNSQYFFS 1351
gi 730615 1407 GD--FDVRSLKDSTYFFS 1422
gi 3914823 1215 GA--FDLEQVKRSTYFFS 1230
|







